A Blog About Computer Aided Design

Creating a 3D part in CATIA
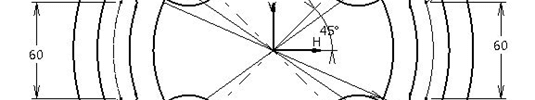
After learning few important commands from Sketcher and Part Design workbench, we can now create a simple 3D part in CATIA. In this post the focus is learning how to use other commands like Hole, Edge Fillet. Here is the 3D object we will try replicating using CATIA. Lets dive right into it

Type of Gears
In technology, the word gear is used to designate a machine element having a purpose of transmitting/receiving power and motion from/to a rotating shaft by means of progressive engagement of their edges called teeth. In mechanical engineering, gears are generally used for power transmission and they are commonly placed in one of the following types:…

Sketcher Workbench in CATIA
This post is to illustrate some important commands and in Sketcher Workbench. You need to remember that a part always starts with a sketch. The simple exercise below will serve as the object of our tutorial. If you are rather an AutoCAD user, you will find these AutoCAD exercises more helpful, them come back and…

Basic commands in CATIA
CATIA (Computer Aided Three-dimensional Interactive Application) is one amongst CAD software that was initially developed to help design aviation systems. It is today one of the leading CAD software in aviation and automobile industry. Here is a very basic “Demo” about Basic commands in CATIA. In this post we will discuss exclusively commands related to…

Easy rendering software
Rendering is one of the most time consuming task while working on a project. Sometime rendering a model can take way too long, and in some case you have to wait for hours just to notice the resulting image of your rendering is not good enough and that you have to make few changes on…

Different CAD views
This post is all about views in technical drawing. This is a reminder, specially for those who will be heading to those AutoCAD 3D tutorials I published here earlier. There is need to remember some basic principles about technical drawing while using any CAD software, here we are all about VIEWS. It is common to…

AutoCAD Trim
Trimming is one of the first tools one should learn how to use while operating on AutoCAD. The AutoCAD TRIM command is a thing you can barely end a project without using. Why should you use the TRIM command in AutoCAD? The TRIM command helps you trim object such to have a new object formed…

AutoCAD 3D Tutorial
One step closer to reality! Looking for some basic AutoCAD 3D exercises? Here are some exercises designed to help you practice and learn how to draw in 3D in AutoCAD. Before diving into this post, make sure you digest what is shown in these 5 3D demo: AutoCAD 3D tutorial 1 AutoCAD 3D tutorial 2…
FreeCAD – AutoCAD alternative
FreeCAD is another AutoCAD alternative in our list. It is a nice 3D modeling software for beginners. It gives you the ability to draw 3D object while giving them parameters thus allowing you to easily come back and modify the modeling parameters of your design after creation. It is a free and open source software.…
LibreCAD – AutoCAD alternative
LibreCAD is another great Alternative to AutoCAD. It is an open source CAD software that will help you create 2D with as much ease as you would do if you were working with AutoCAD. In my opinion this is a pretty nice alternative for people just starting and this will also be a new experience…

Steering Knuckle
A steering knuckle is one of the most important component in automotive suspension system. This rather odd looking mechanism coordinates the steering mechanism of a vehicle with the suspension system and then the actually wheel. Depending on the vehicle and suspension design, the steering hub or spindle will also vary slightly. In the case of…

Using the command window in AutoCAD
The command window in AutoCAD, for the less I can say, is where all what you are doing is being recorded. It is also the means AutoCAD uses to communicate with you. It is important to know how to use it. All icons are shortcut of commands. let’s take as an example the process of…

How to draw a spring in AutoCAD
Drawing a spring in AutoCAD is relatively easy using the SWEEP command. If you had a look at how I started working on AutoCAD 3D you would know a little about the SWEEP command. But before getting to using the SWEEP command, I would like to go through a simple but primitive way to easily…

UCS in AutoCAD
UCS stands for User Coordinate System and is represented by the object on the left down corner of your AutoCAD windows. This is a crucial tool for 3D design in AutoCAD. This is why in this post I will use the simple figure below to help you have an idea about how mastering UCS will…

Basic Tools in 3D in AutoCAD
While a lot will give you many reason why AutoCAD is not the indicated software for 3D I want to deliver in this post the concept of 3D in AutoCAD and what you should be thinking about if 3D is a total new concept for you. 3D stand for 3dimensions right? so the most basic…

Best Free CAD Software for Beginners
Free CAD software for beginners is the topic we will discuss today. As the title states, this is not an article for professional graphic designers but one for the budding architect, the prospective engineer, and the aspiring graphic designer planning to make a name in his or her chosen profession. Knowing what the best CAD…

The Best Free AutoCAD Alternative
While running the last series of 2D AutoCAD exercises, I felt it would be unfair to those who wanted to learn CAD but did not own the AutoCAD software. I am choosing to begin this set of experiments with DraftSight, which I recently installed and in my opinion is one of the best free and…

Annotation scaling in AutoCAD
Here is how to work with Annotation scaling in AutoCAD Sometime, annotations might be either too big or too small compared to dimensions you used in your project. Here is how you can change annotation features in AutoCAD, including text, and color The DIMSTYLE command help you access the DIMENSION STYLE MANAGER which you can…

How to link an Excel file to AutoCAD
Here is a quick way How to link an Excel file to AutoCAD. The TABLE command in AutoCAD allows you not to only import your data from excel to AutoCAD but it also allows you to link both files together. That will make any change from excel to be having a possibility to being updated…

How to write text on arc in AutoCAD
Here is how to write text on arc in AutoCAD in 3 simple steps. Step 1 Draw your ARC, using a circle and trimming it or using the ARC command. Step 2 Go on the EXPRESS TOOLS tab, click on ARC ALIGNED TEXT and select the ARC you created in step 1
















Got any book recommendations?